Co je Marfanův syndrom?
Marfanův syndrom popisuje komplexní dědičnou poruchu pojivové tkáně, která postihuje především oči, kardiovaskulární systém a systém kosterního svalstva. Vzhledem k tomu, že každý orgán je tvořen pojivovou tkání, může Marfanův syndrom ideálně zničit a silně narušit růst a funkci každého anatomického místa.
Syndrom je přenášen jako autozomálně dominantní rys: proto jsme konfrontováni se závažným genetickým onemocněním, které má extrémně variabilní fenotypovou expresi (defekty se mohou výrazně lišit od rodiny k rodině nebo od pacienta k pacientovi).
Co způsobuje Marfanův syndrom, je změna genu FBN1 (na chromozomu 15), který kóduje fibrilin-1, velmi důležitý pojivový glykoprotein, který představuje strukturní podporu pro mikrofibrily.
Mikrofibrily: skládající se z fibrilinu, jsou přítomny v extracelulární matrici, ve které tvoří tkaninu pro ukládání elastinu v elastických vláknech. Ačkoli všudypřítomné v těle, mikrofibrily oplývají hlavně v aortě, vazech a zonulích řasnatých těl (na úrovni očí).
Jako autosomálně dominantní onemocnění jsou u Marfanova syndromu postiženy pouze děti, které zdědily gen FBN-1 změněný oběma rodiči. Nicméně v jednom případě ze 4 je onemocnění výsledkem spontánních mutací u pacientů, kteří nemají rodinnou anamnézu.
Název onemocnění pochází od francouzského pediatra, který jej poprvé popsal v roce 1896 (A. Marfan), po kterém musel počkat až do roku 1991, aby identifikoval změněný gen, který se podílel na symptomatickém projevu: objevitelem byl F. Ramirez.
Podívejte se na video
X Podívejte se na video na youtubepříčiny
Zmínili jsme se, že Marfanův syndrom je okamžitým vyjádřením mutace genu, který kóduje fibrilin-1. Pokusme se toto téma prohloubit, abychom pochopili, který mechanismus je založen na spuštění tohoto syndromu.
FIBRILLINA 1 je glykoproteinová složka elastinu, nezbytná pro zajištění a udržení pružnosti a pevnosti tkáně. Za fyziologických podmínek se fibrilin 1 váže na jiný protein, známý jako TGF-beta (nebo faktor faktoru transformujícího růstu). Zdá se, že TGF-beta se podílí na škodlivých procesech ovlivňujících vaskulární hladký sval a extracelulární matrix. Vycházeje z těchto předpokladů, někteří autoři jsou přesvědčení, že Marfanův syndrom je způsoben, kromě mutace genu FBN-1, také přebytkem TGF-beta, zejména v aortě, v srdečních chlopních a v plicích.
výskyt
Odhaduje se, že Marfanův syndrom postihuje 1 subjekt každých 3 000 až 5 000 porodů a projevuje se bez rozdílu mezi muži a ženami, bez předurčení pro přesné plemeno. Statistiky ukazují, že 75% pacientů má pozitivní rodinnou anamnézu; ve zbývajících 25% příčina spočívá v sporadických mutacích, které se zdají být nějakým způsobem spojeny s pokročilým věkem otce v době početí.
Děti s extrémně těžkými formami Marfanova syndromu mají délku života kratší než jeden rok.
Před vývojem operačních strategií otevřené srdce měla většina pacientů s Marfanovým syndromem průměrnou délku života 32 let; díky neustálému zlepšování lékařských a farmakologických terapií žijí pacienti s Marfanovým syndromem v průměru až 60 let.
Příznaky a příznaky
Další informace: Symptomy Marfanův syndrom
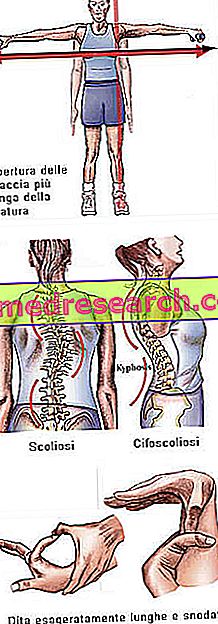
Marfanův syndrom může běžet zcela asymptomaticky. Postižení pacienti mají nadměrně štíhlou strukturu, která je nepřiměřeně vysoká a tenká. Dolní a horní končetiny mají mnohem vyšší délku než kmen (dolicostenomegalia). Tam je také mluvit o arachnodactyly nejlépe vyjádřit pojetí přehnané délky prstů, typický pro osoby postižené Marfan syndromem: ruce jsou proto srovnávány s nohama pavouka.
Pokud jde o výšku, tito pacienti mají průměrnou výšku nad 97. percentilem.
Mezi další charakteristické rysy, které se často vyskytují u pacientů s Marfanovým syndromem, patří:
- Ramena otevřená větší než výška
- Volné klouby → přehnaná pohyblivost kloubů
- Deformace hrudní stěny
- Krystalické přemístění
- Horní část těla je méně rozvinutá než spodní část
- Spontánní pneumotorax (11%)
- skolióza
- Kožní strie na stehně, zádech, deltoidní, prsní úrovni
Mezi nejproblematičtějšími příznaky spojenými s Marfanovým syndromem je prolaps srdeční chlopně a insuficience mitrální chlopně: podobný stav může snadno podpořit dilataci aortálního prstence a aortální disekce.
Tabulka ukazuje příznaky, které lze nalézt u pacientů s Marfanovým syndromem. Zde popsané znaky nejsou vždy přítomny, ale dobrá část z nich může být nalezena.
| Postižené anatomické místo | Možné příznaky |
roztomilý | Striae v hrudní, bederní a sakrální oblasti |
oči | Poruchy vidění, astigmatismus, odchlípení sítnice, glaukom s úzkým úhlem, dislokace čočky, krátkozrakost |
Kostní struktura | Artralgie, kyfoskolióza, dolikostenomelie (nadměrný vývoj délky končetin s ohledem na trup), hypermobilita, vysoký patra, deformovaný hrudník, ploché nohy, úzké a tenké zápěstí, abnormální opakovaný vstup / výstup hrudní kosti, skolióza, zakřivená ramena, spondylolistéza |
Dita | arachnodactyly |
plíce | Spontánní pneumotorax, dušnost, idiopatická plicní obstrukční choroba |
Změny obličeje | Ogival patra (malformace patra), mandibulární retrognathia (vada ve vývoji čelisti), prodloužená tvář |
srdeční | Angina pectoris, aneuryzma břišní aorty, srdeční arytmie, dilatace / prasknutí / disekce hrudní aorty, aortální insuficience, prolaps mitrální chlopně |
jazyk | Jazyková obtížnost |
diagnóza
Vzhledem k více než 200 možným mutacím je použití genetických markerů pro diagnostické účely téměř nemožné.
Zjištění Marfanova syndromu není vždy tak bezprostřední, vzhledem k tomu, že fenotypová exprese mutace není vždy zřejmá a snadno identifikovatelná. Diagnostické zpoždění může vážně ohrozit přežití pacienta: stačí si například uvědomit, že nedošlo k rozpoznání kardiovaskulárního problému.
Diagnostická kritéria pro Marfanův syndrom byla vypracována mezinárodně v roce 1996: diagnóza spočívá ve zkoumání rodinné historie spojené s kombinací hlavních a vedlejších ukazatelů syndromu.
Některé z mnoha diagnostických testů jsou:
- echocardiogram
- Magnetická angioisonance a CT (pro výzkum aorty)
- magnetická rezonanční angiografie (MRA) s kontrastní tekutinou (aby se vnitřní struktury aorty projevily)
- vyšetření se štěrbinovými lampami (pro analýzu případné dislokace čočky)
- měření očního tlaku (zdůraznění možné přítomnosti glaukomu)
- genetické testy (doporučeno před počátkem dítěte, aby se zjistil syndrom nebo ne)
terapie
Být genetickou nemocí, neexistuje žádný lék nebo léčba, která by mohla zvrátit nemoc.
Použití léků je však nezbytné pro zmírnění symptomů a zamezení jakýchkoli komplikací, zejména kardiálních. Pro tento účel jsou zvláště vhodné léky na snížení krevního tlaku, jako jsou sartany (zejména), inhibitory ACE a beta-blokátory.
V souvislosti s Marfanovým syndromem mohou pacienti trpící skoliózou sledovat i specifickou péči, stejně jako pacienti s glaukomem.
Chirurgie je možná k nápravě anomální dilatace aorty, prvku, který často sjednocuje většinu pacientů s Marfanovým syndromem.
Pokračovat: Marfanův syndrom - léky a péče »