všeobecnost
Kraniosynostóza je termín, kterým lékaři indikují abnormalitu lebky v důsledku předčasné fúze jednoho nebo více lebečních stehů.
Kraniální švy jsou vláknité klouby, které spojují kosti kraniální klenby (tj. Frontální, temporální, parietální a týlní kosti).
Kraniosynostóza může být izolovaným jevem (nesyndromická kraniosynostóza) nebo výsledkem některých morbidních stavů (syndromická kraniosynostóza). Mezi morbidní stavy, které způsobují předčasnou fúzi kraniálních stehů, jsou nejznámější: Crouzonův syndrom a Apertův syndrom.
S předčasnou fúzí lebečních stehů nemají mozkové struktury dostatečný prostor pro růst. To má různé důsledky, zejména zvýšení intrakraniálního tlaku (intrakraniální hypertenze).
Rychlá a přesná diagnóza umožňuje naplánovat ad hoc léčbu. Ten je chirurgického typu a jeho konečným cílem je včasné oddělení šicích stehů.
Vzpomínky na anatomii lidské lebky
Lebka, vybavená kostmi a chrupavkami, je kosterní strukturou hlavy, která tvoří obličej a chrání mozek, mozeček, mozek a smyslové orgány.

Pro zjednodušení studia a pochopení lebky si anatomové mysleli, že ji rozdělí na dva oddíly, nazývané neurokranium a splancnocranium .
neurocranium
Neurokranium je horní lebeční oblast, obsahující mozek a některé z hlavních smyslových orgánů. Nejdůležitějšími kostmi - striktně plochými - jsou frontální, temporální, parietální a týlní kosti; tyto dohromady tvoří tzv. lebeční klenbu .
splanchnokraniu
Splanchocranium, neboli obličejový masiv, je antero-nižší oblast lebky, složená z rovných a nerovných kostí. Představuje kosterní strukturu obličeje, proto obsahuje kostní prvky, jako je čelist, horní čelist, lícní kosti, nosní kost atd.
Co je kraniosynostóza?
Kraniosynostóza je vzácná anomálie lebky, charakterizovaná nepřirozeným tvarem hlavy v důsledku předčasné fúze jednoho nebo více lebečních stehů . Kraniální švy jsou vláknité klouby, které spojují kosti kraniální klenby (tj. Frontální, temporální, parietální a týlní kosti).

Z webu: //www.wkomsi.com/
KDY BY MĚLI BÝT ZAVŘENO UZAVŘENÍ ZÁVĚSŮ?
Za normálních podmínek dochází k fúzi lebečních stehů v postnatálním období (Pozn .: některé procesy dokonce končí ve věku 20 let). Tento dlouhý proces fúze umožňuje mozku růst a rozvíjet se správně.
Pokud, stejně jako v případě kraniosynostózy, dochází k fúzi příliš brzy - tedy během prenatálního, perinatálního * nebo raného dětství - encefalické elementy (mozek, mozeček a mozkový mozek) a některé smyslové orgány (zejména oči) procházejí změna tvaru a růstu.
příčiny
Patofyziologický proces, který určuje kraniosynostózu, je předčasná fúze lebečních stehů .
Tento proces může představovat izolovaný jev - kde „izolovaným“ myslíme, že není spojen s žádným konkrétním morbidním stavem - nebo může být důsledkem některých konkrétních syndromů, téměř vždy genetické povahy.
Ve světle toho se lékaři rozhodli klasifikovat kraniosynostózu do dvou kategorií:
- Syndromová kraniosynostóza . Termín nesyndromní znamená, že kraniální anomálie není spojena s žádnou patologií nebo jinou fyzickou vadou.
- Syndromická kraniosynostóza . Termín syndromní znamená, že kraniální malformace je výsledkem určitého syndromu, ve většině případů genetického typu.
NEININDROMICKÁ CRANIOSINOSTÓZA
Lékaři a výzkumníci dosud nezjistili, jaké jsou příčiny nesyndromické kraniosynostózy.
Navrhli různé hypotézy - včetně vlivu environmentálních faktorů nebo problémů hormonálního typu - ale žádná z těchto teorií nepotvrdila experimentální výsledky.
Abychom pochopili přesný původ této anomálie, jsou v tomto ohledu zapotřebí další studie.
CRANIOSINOSTOSI SINDROMICA
Podle nejnovějšího lékařského výzkumu existuje více než 150 různých syndromů, z nichž všechny jsou vzácné a způsobují kraniosynostózu.
Mezi těmito syndromy jsou nejznámější a nejčastější:
- Crouzonův syndrom . Výsledek specifických mutací v genech FGFR2 (chromozom 10) a FGFR3 (chromozóm 4), tento konkrétní morbidní stav postihuje jednoho novorozence každých 60 000 a vede k přítomnosti anomálií výhradně na úrovni hlavy a obličeje.
- Apertův syndrom . Vyplývá hlavně z mutací genu FGFR2 (stejně jako u Crouzonova syndromu) a postihuje jednoho novorozence každých 100 000.
Na rozdíl od Crouzonova syndromu jsou genetické změny FGFR2 takové, že malformace zahrnují nejen lebku a obličej, ale také ruce a nohy.
- Pfeifferův syndrom . Vzniká v důsledku mutací "obvyklého" genu FGFR2 a genu s podobnými funkcemi, nazývaného FGFR1 (chromozóm 8). Zvláštností těchto mutací je, že - kromě deformit lebky a obličeje - také určují: syndaktyly, brachydaktyly a palce a velké prsty (disproporcionální vůči ostatním prstům).
Pfeifferův syndrom má výskyt jednoho případu na 100 000 novorozenců.
- Saethre-Chotzenův syndrom . Je to genetický stav, který postihuje novorozence každých 50 000 nebo tak. Způsobuje různé malformace lebky, obličeje, rukou a nohou. Některé specifické mutace genu TWIST1, umístěné na chromozomu 7, jsou zodpovědné za nástup Saethre-Chotzenova syndromu.
EPIDEMIOLOGIE CRANIOSINOSTOSI
Na základě nejnovějších statistik se zdá, že dítě trpí kraniosynostózou každých 1800-3000.
Pokud jde o nejvíce postižené pohlaví, několik klinických studií prokázalo, že 3 ze 4 pacientů jsou muži. Důvod, proč je kraniosynostóza v mužské populaci rozšířenější, je zcela neznámý.
Rizikové faktory kraniosynostózy.
- Nízká porodní hmotnost
- Předčasný porod
- Pokročilý věk otce
- Kouření matky během těhotenství
Příznaky a komplikace
Většina symptomů pozorovaných v přítomnosti kraniosynostózy je způsobena zvýšeným tlakem uvnitř lebky . V medicíně se zvýšení tlaku uvnitř lebky nazývá intrakraniální hypertenze nebo intrakraniální hypertenze .
V přítomnosti kraniosynostózy je intrakraniální hypertenze důsledkem skutečnosti, že mozek a další struktury uvnitř lebky nemají správný prostor k růstu, takže jdou tlačit na kostní struktury hlavy.
Je třeba si uvědomit, že je důležité si uvědomit, že pokud jsou kraniální stehy mnoho nebo pokud stav není léčen včas, kraniosynostóza může vést ke sníženému rozvoji kognitivních schopností a nízkému IQ.
Příznaky ENDOCRANIC HYPERTENSION
Možné příznaky intrakraniální hypertenze jsou:
- Trvalé bolesti hlavy. Obecně se zhoršuje ráno a v noci.
- Problémy s viděním. Jedná se o dvojité vidění, rozmazané vidění a rozmazané vidění.
- zvracení
- popudlivost
- Nadýchané nebo prominentní oči
- Obtížnost při sledování pohybu předmětů
- Problémy se sluchem
- Respirační problémy
- Změny duševního stavu
- papilledema
Počet kraniálních stehů zapojených do vývoje kraniosynostózy má rozhodující vliv na přítomnost intrakraniální hypertenze.
Lékaři například pozorovali, že účast jediného kraniálního švu vyvolává intrakraniální hypertenzi u 15% pacientů; zatímco zapojení alespoň dvou stehů vede ke zvýšení tlaku uvnitř lebky u nejméně 60% pacientů.
V přítomnosti mírné formy kraniosynostózy začíná být intrakraniální hypertenze problematická, což způsobuje výše uvedenou symptomatologii, kolem 4-8 let života.
ZNAKY CRANIOSINOSTOSI
Mezi příznaky kraniosynostózy patří nejčastější:- Formace tuhých hřebenů podél lebečních stehů
- Abnormality kraniálních fontanel
- Hlava s rozměry není přiměřená zbytku těla
TYPY CRANIOSINOSTÓZY
Tvar hlavy pacientů s kraniosynostózou závisí na tom, které kraniální stehy se předčasně uzavřely.
Po pozorování to lékaři považovali za vhodné rozlišovat kraniosynostózu v různých typech, v závislosti na použitých kraniálních šicích.
Typy kraniosynostózy jsou:
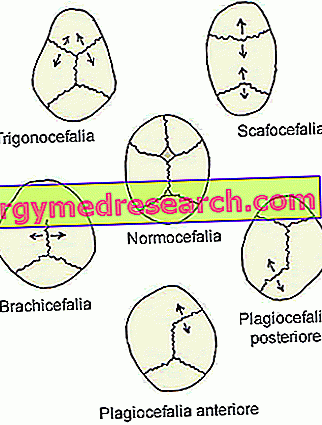
- Sagitální synostóza ( dolichocephaly nebo scaphocephaly ). Je to nejběžnější typ kraniosynostózy; ve skutečnosti charakterizuje asi polovinu klinických případů.
Jeho přítomnost se shoduje s předčasným uzavřením sagitálních lebečních stehů, umístěných v horní části lebky, mezi parietálními kostmi.
Od //en.wikipedia.org/wiki/Plagiocephaly
- Koronální kraniosynostóza ( brachycefalie ) Je to druhý nejčastější typ kraniosynostózy; charakterizuje jeden klinický případ každé čtyři.
Jeho nástup zahrnuje předčasnou fúzi koronálních stehů, které probíhají mezi frontální kostí a parietálními kostmi.
- Metopická synostóza ( trigonocefalie ). Jedná se o velmi vzácný typ kraniosynostózy, která rozlišuje pouze 4-10% případů.
Jeho vzhled se shoduje s předčasnou fúzí metopického (nebo frontálního) stehu, který probíhá od nosu k horní části hlavy, oddělující frontální kost ve dvou. Obecně se tento šev přirozeně osidluje v šestém roce života.
- Lambdoidní synostóza ( plagiocephaly ). Je to nejvzácnější typ kraniosynostózy. Ve skutečnosti je to pouze 2-4% klinických případů.
Jeho přítomnost zahrnuje časnou fúzi lambdoidního stehu, umístěného mezi parietálními kostmi a týlní kostí, v zadní části hlavy.

KOMPLIKACE
Kromě narušení intelektuálního vývoje může neléčená kraniosynostóza určit:
- Takzvaný obstrukční syndrom spánkové apnoe .
- Trvalé změny obličeje, zejména v očích a uších.
- Trvalé deformity na základně lebky (například malformace nebo syndrom Arnold-Chiari).
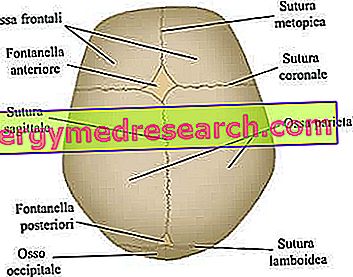
Hlavní kraniální švy zapojené do procesu kraniosynostózy. Z webu: www.sciencebasedmedicine.org
- Hydrocefalus .

diagnóza
Pro diagnostiku kraniosynostózy jsou nezbytné fyzikální vyšetření, vyhodnocení klinické anamnézy a radiologické snímky poskytované rentgenovými paprsky nebo CT skenováním do hlavy .
Pokud je kraniosynostóza syndromního typu, je také důležité stanovit morbidní stav, který vedl k jeho nástupu. Lékaři by se proto mohli uchýlit k krevním testům a především k genetickému poradenství .
CÍL ZKOUŠKY
Fyzikální vyšetření spočívá v pečlivé analýze klinických příznaků přítomných na hlavě subjektu, podezřelých z kraniosynostózy, lékařem.
Pediatr je obecně odpovědný za provádění této důležité diagnostické kontroly.
KLINICKÁ HISTORIE
Vyhodnocení klinické anamnézy je důležité pro diagnostické účely, neboť zahrnuje otázky týkající se rizikových faktorů kraniosynostózy.
Lékař (obvykle vždy pediatr) proto prozkoumá, zda:
- Dítě se narodí předčasně nebo má nízkou hmotnost.
- Jaký byl věk otce v době početí.
- Pokud matka kouří během těhotenství.
RADIOLOGICKÉ ZKOUŠKY
Rentgenové paprsky a CT v hlavě slouží k potvrzení diagnózy více než cokoliv jiného a ukázat lékaři, které kraniální stehy se předčasně roztavily.
Znalosti, o které se jedná, umožňují plánování nejvhodnější chirurgické léčby.
léčba
Kraniosynostóza je léčitelná pouze chirurgickým zákrokem .
Ta spočívá v operaci separace kraniálních stehů předběžně fúzujících jeden od druhého.
Konečným terapeutickým cílem chirurgie je poskytnout encefalickým strukturám a některým smyslovým orgánům, například očím, prostor, který je nezbytný k tomu, aby se vyvíjely a fungovaly v nejlepším smyslu.
NEJLEPŠÍ ČAS DO INTERVENE
Neexistuje žádná celková dohoda ze strany lékařů o tom, co je nejlepší doba k provedení operace kraniosynostózy.
Podle některých odborníků by ideální období pro operaci bylo v pozdním dětství, kdy riziko recidivy je nižší (tj. Druhá předčasná fúze lebečních stehů). V případě relapsu se operace musí ve skutečnosti opakovat a vzhledem k laskavosti procedury se nedoporučuje.
Podle jiných odborníků by nejvhodnější čas byl v raném dětství (mezi 6 a 12 měsíci života), kdy lebka ještě není zcela osifikovaná a kosti jsou stále tvarovatelné. Možnost tvarování kostí (tvárnost) umožňuje, aby byly vyřešeny jakékoliv morfologické abnormality kostí, což by mohlo způsobit závažné estetické defekty a funkční problémy (do čelisti nebo očí) ve zralém věku.
MOŽNÉ CHRÁNĚNÉ PŘÍSTUPY
Existují dva různé chirurgické postupy: tradiční chirurgie, nazývaná také „otevřená“ a endoskopická chirurgie.
- Tradiční (nebo "otevřená") operace .
Předpokládá celkovou anestezii (pacient je tedy v průběhu operace zcela v bezvědomí) a praxi chirurgického řezu na hlavě, přesně v místě, kde radiologické snímky ukázaly kraniální anomálii.
Operační chirurg (neurochirurg) odstraní abnormální kost přes řez na hlavě a svěří ji specialistovi na kraniofaciální chirurgii, který ji modifikuje a dává jí tvar, který umožňuje normální vývoj mozkových struktur.
Po úpravě neurochirurg znovu vloží kost do původní polohy a uzavře incizi stehy.
Stejně jako mnoho tradičních operací je i „otevřená“ operace poněkud invazivní; nicméně, aby to bylo výhodné, je skutečnost, že je schopna přesně modifikovat strukturu kosti a s dobrými výsledky.
- Operace endoskopické chirurgie .
Zahrnuje použití endoskopu, nástroje podobného pružné trubce, vybavené optickou kamerou na jednom konci a připojené k monitoru.
Z provozního hlediska spočívá v zasunutí endoskopu do otvoru vytvořeného na lebce a v oddělení, pomocí samotného endoskopu, předběžně.
Neurochirurg zvládá orientaci v hlavě díky obrazům, které kamera promítá na externě připojený monitor.
Endoskopická chirurgická intervence je rozhodně méně invazivní než „otevřená“ operace (doba hospitalizace je také kratší), má však dvě nevýhody: je indikována pouze pro pacienty po dobu několika měsíců (6 obecně), které mají tvarovatelné kosti; je vyšší riziko opakování.
POST-OPERATIVNÍ FÁZA
Obecně platí, že pacient s kraniosynostózou, podrobený chirurgickému zákroku, musí zůstat v nemocnici po dobu asi 4-5 dnů po operaci. Během této doby neurochirurg a jeho zaměstnanci pravidelně sledují jejich vitální funkce a kontrolují, zda všechno funguje dobře.
Po rezignaci je naplánována série pravidelných kontrol, které jsou nejprve, půl roku a pak, s růstem pacienta ročně.
prognóza
Prognóza závisí na různých faktorech, včetně:
- Příčiny, které vyvolaly kraniosynostózu. Některé genetické nemoci zodpovědné za tuto anomálii jsou velmi závažné a mají špatnou prognózu.
- Pozice stehů se brzy spojila. Pokud jsou stehy umístěny v polohách, které jsou pro neurochirurga "nepříjemné" k dosažení, zásah kraniosynostózy se stává komplikovanou a nemusí poskytovat požadované výsledky.


