všeobecnost
Cystická fibróza je nejčastější autosomálně recesivní onemocnění u kavkazské populace, které postihuje přibližně 1 jedince v každé 2500.
Tento patologický stav je znám pro své škodlivé účinky na dýchací systém, ale ovlivňuje i jiné systémy, jako jsou trávicí a reprodukční systémy.
U jedinců s cystickou fibrózou jsou dýchací cesty ucpány hustým, viskózním hlenem, který je obtížné odstranit i při nejúčinnějším kašli. Dýchání se stává obtížným a pacienti - pokud se nepřetržité úsilí nevynakládá na udržení čistoty dýchacích cest několikrát denně - hrozí riziko úmrtí v důsledku vlastní sekrece. Pacienti s cystickou fibrózou umírají často v důsledku pneumonie, protože ucpané dýchací cesty poskytují úrodné prostředí pro růst bakterií.

příčiny
Cystická fibróza je způsobena mutacemi v transmembránové vodivosti genu cystické fibrózy (CFTR) lokalizované na chromozomu 7 (mapování lokusu: 7q31).
Je známo alespoň 1500 mutací genu CFTR. Nejčastější mutace se běžně nazývá "Delta-F508" (DF508) a je určena delecí 3 párů bází v exonu 10, což vede ke ztrátě fenylalaninu v poloze 508.
Protein kódovaný CFTR genem je transmembránový kanál patřící do ATPázové superrodiny transportních nebo ABC transportérů, který se nachází na úrovni apikální membrány epitelových buněk a je závislý na transportu iontů chloru.
Za normálních podmínek určité buňky, které lemují dýchací cesty, vylučují hlen spolu s vodnou kapalinou, která snižuje jejich hustotu. U cystické fibrózy je vylučování vodné tekutiny značně sníženo , v důsledku čehož se hlen stává velmi hustým a obtížně odstranitelným z dýchacího traktu.
V epitelu dýchacích cest, stejně jako všechny epitely, které nesou tekutiny, závisí transport vody na transportu rozpuštěných látek. Aby se vyloučila voda, buňky dýchacího epitelu aktivně transportují ionty chloru (Cl-) z intersticiální tekutiny do lumenu, což vytváří negativní elektrický potenciál, který způsobuje pasivní tok sodíku (Na +) ve stejném směru. Pohyby Na + a Cl- zvyšují osmotický tlak kapaliny, která zvlhčuje sklon epitelu otočený směrem k lumenu, v důsledku čehož se voda pohybuje pasivně podle osmotického gradientu, z intersticiální kapaliny do lumenu. Genový defekt, který ovlivňuje nástup cystické fibrózy přímo zabraňuje transportu Cl- a nepřímo interferuje s transportem Na + a vody. V epitelu tedy není vytvořen osmotický gradient nezbytný pro vylučování vody.
Rizikové faktory
- Rodinné dědictví . Vzhledem k tomu, že cystická fibróza je dědičné onemocnění, které se přenáší autosomálně recesivním způsobem, je důležité zvážit rodinnou anamnézu (budoucí anamnézu) budoucích rodičů.
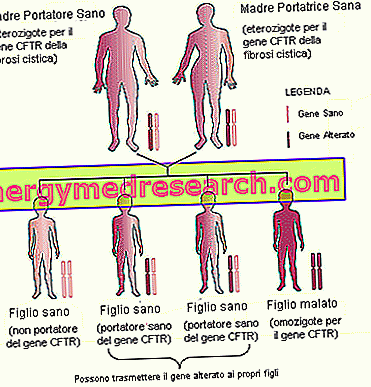
- Plemeno . Výskyt cystické fibrózy je větší u bílých lidí na severu a evropského původu.

Příznaky a klinické příznaky
Další informace: Příznaky cystické fibrózy
Závažnost symptomů se může lišit v závislosti na průběhu onemocnění: většina klinických příznaků ovlivňuje dýchací systém a gastrointestinální systém.
Dýchací příznaky a symptomy
Silný a viskózní hlen spojený s cystickou fibrózou způsobuje obstrukci dýchacích cest a inhibuje aktivitu řasinky, která lemuje plicní epitel, což usnadňuje eliminaci samotného hlenu. V malých dýchacích cestách výsledná stagnace sliznic přispívá k vytvoření ideálního prostředí pro bakterie, jako jsou Streptococcus aureus nebo Pseudomonas aeruginosa, které předurčují postižené osoby k opakovaným infekcím a zánětům, které mohou být také závažné, jako pneumonie. Chronická povaha těchto zánětlivých procesů může časem poškodit plicní tkáň a následně vést ke špatným dýchacím funkcím.
Charakteristické symptomy, které se vyvíjejí v průběhu cystické fibrózy, zahrnují:
- Trvalý kašel s produkcí sputa
- Dech
- Dušnost a potíže s dýcháním
- Rekurentní plicní infekce
Příznaky a příznaky trávicího traktu
Tlustý hlen může také okludovat kanály, které přenášejí trávicí enzymy z pankreatu do tenkého střeva. Bez těchto trávicích enzymů, střevo nemůže strávit a úplně absorbovat živiny v přijaté potravě (obzvláště tuky, cholesterol a vitamíny rozpustné v tucích, A, D, E a K).
Výsledkem je často následující:
- Oteklé břicho, meteorismus, steatorrhea
- Špatný přírůstek hmotnosti a špatný růst (v důsledku podvýživy)
- Střevní blokáda, zejména u novorozenců (meconium ileum)
- Zácpa nebo těžká zácpa
komplikace
Komplikace dýchacího systému
- Bronchiektasie : cystická fibróza je jednou z hlavních příčin bronchiektázie, což je stav, který poškozuje dýchací cesty, což má za následek obstrukční ventilační selhání, s výrazným snížením výdechového průtoku.
- Chronické infekce : tlustý hlen, který stagnuje v plicích a dutinách, nabízí ideální živnou půdu pro bakterie a houby. Jako výsledek, lidé s cystickou fibrózou mohou mít časté ataky sinusitidy, bronchitidy nebo pneumonie.
- Nosní polypy : protože vnitřní výstelka nosu je zanícená a oteklá, mohou se vyvíjet měkké masité výrůstky (polypy), které mohou bránit dýchání během spánku.
- Kašel krví (hemoptýza): v průběhu času může cystická fibróza způsobit ztenčení stěn dýchacích cest. V důsledku toho mohou pacienti s cystickou fibrózou emitovat krev z dýchacích cest, obvykle kašlem.
- Pneumothorax : tento stav, který spočívá v hromadění vzduchu v pleurální dutině, je častější u starších lidí s cystickou fibrózou. Pneumothorax může způsobit bolest na hrudi a dušnost.
- Kolaps plic : chronické plicní infekce mohou poškodit plicní parenchymu, což způsobuje pravděpodobnější zhroucení plic.
- Respirační insuficience : v průběhu času může cystická fibróza poškodit plicní tkáň a ohrozit její funkčnost.
Komplikace trávicí soustavy
- Nutriční nedostatky : tlustý hlen může blokovat pankreatické kanály, které přenášejí trávicí enzymy z pankreatu do tenkého střeva. Bez těchto enzymů nemůže tělo absorbovat proteiny, tuky a vitaminy rozpustné v tucích.
- Diabetes : pankreatické beta buňky vylučují inzulín, který je nezbytný pro zprostředkování regulace glykémie. Cystická fibróza zvyšuje riziko vzniku diabetu. Přibližně 20% postižených se rozvine ve věku 30 let, takže pacienti jsou pravidelně testováni na krevní glukózu.
- Obstrukce žlučovodů : kanál, který nese žluči z jater a ze žlučníku do tenkého střeva, může být blokován a zapálen, což vede k problémům s játry a někdy žlučovým kamenům.
- Rektální prolaps : častý kašel nebo námaha během defekace (zácpa) může způsobit vyčnívání vnitřní rektální tkáně z řitního otvoru.
- Intususcepce : děti s cystickou fibrózou jsou vystaveny vyššímu riziku intususcepce, což je stav, kdy se část střevních záhybů ohýbá zpět na sebe a vytváří invaginace. Výsledkem je střevní obstrukce, nouzová situace, která vyžaduje okamžitou léčbu.
Komplikace reprodukčního systému
Téměř všichni muži s cystickou fibrózou jsou sterilní, protože kanály, které spojují varlata a prostatickou žlázu (vas deferens), jsou blokovány hlenem nebo zcela chybí (nepřítomnost kanálků je stále neznámá příčina).
Ačkoli ženy s cystickou fibrózou mohou být méně plodné než jiné ženy, stále si zachovávají schopnost otěhotnět a ukončit těhotenství. Gestace však může přispět ke zhoršení symptomů cystické fibrózy.
Další komplikace
Osteoporóza : osoby s cystickou fibrózou jsou vystaveny vyššímu riziku vzniku osteoporózy s následnou ztrátou hmotnosti a síly kostí. Pacienti by měli být poučeni, aby v případě potřeby užívali vápník, vitamin D a bisfosfonáty.
Nerovnováha elektrolytů : lidé s cystickou fibrózou mají v potu vyšší než normální hladinu soli. Příznaky a symptomy, jako je zvýšení srdeční frekvence, únava, slabost a nízký krevní tlak, pomáhají zdůraznit dekompenzaci, kterou podstupuje hydro-fyziologická rovnováha v krvi.
Diagnostika a léčba cystické fibrózy »


